- Visibility 349 Views
- Downloads 33 Downloads
- DOI 10.18231/j.ijceo.2025.007
-
CrossMark

A study on ocular and systemic manifestations of retinitis pigmentosa
- Author Details:
-
 Rakshanda Hariharan *
Rakshanda Hariharan *
-
Satyanarayana Reddy Madduru
-
Ugandhar Reddy Bommana
Introduction
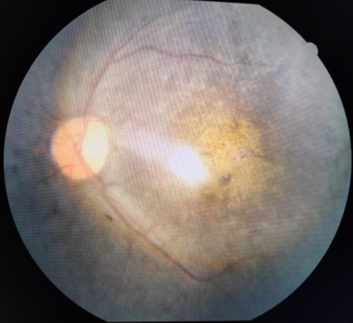
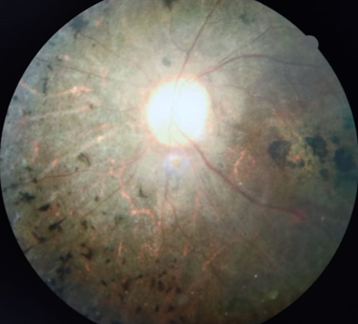
The term “retinitis pigmentosa” was first coined by the famous Dutch ophthalmologist F.C. Donders in 1857. Retinitis pigmentosa (RP) is a group of hereditary disorders of the photoreceptors and retinal pigment epithelium (RPE) which gradually causes night blindness and progressive constriction of the visual field, waxy optic disc pallor, arteriolar attenuation are found in almost all cases. The pigmentary changes seen are characteristic of bone spicules, diffuse granularity or stippling and pigment clumping; these are due to photoreceptors degeneration, atrophy in the outer retina and pigment epithelium.[1] ([Figure 6]) The conservative global prevalence estimate for RP is 1 in 4,000. The inheritance patterns in RP are autosomal dominant, autosomal recessive, X-linked recessive or isolated. The age of onset and severity vary according on the inheritance pattern. Autosomal dominant RP provides a milder clinical picture, with adequate central vision lasting into the sixth decade. The most severe and least prevalent in India is the X-linked recessive type, which causes significant visual impairment in males by the fourth decade.[2]
The clinical diagnosis is based on the presence of night blindness and peripheral visual field abnormalities, fundus lesions, hypovolted electroretinogram traces, and progressive worsening of these symptoms. Molecular diagnosis is possible for some genes, although it is rarely conducted due to the disease's extreme genetic variability. Currently, there is no medicine that can stop the progression of the disease or restore eyesight, so the visual prognosis is low. The therapeutic strategy is limited to slowing the degenerative process with solar protection and vitaminotherapy, treating complications (cataract and macular edema), and assisting patients in dealing with the social and psychological consequences of blindness. However, new therapeutic techniques are emerging through extensive study (genetherapy, neuroprotection, retinal prosthesis). In this study we aim to study the pattern of ocular and systemic manifestations associated with retinitis pigmentosa and syndromic correlation and preventive strategies of the same.
Materials and Methods
This is a prospective study done on patients visiting ophthalmology out patient department at Government Regional Eye Hospital, Kurnool for visual handicap certification, over a period of 1 year. The data includes each patient’s, age, sex, complaints, systemic examination findings, visual acuity, IOP, anterior segment findings, dilated fundus examination, B scan ultrasonography and visual field analysis.
Inclusion criteria
Typical retinitis pigmentosa
Syndromic retinitis pigmentosa
Exclusion criteria
Sector retinitis pigmentosa
RP sine pigmentosa
Retinitis punctata albescens
Secondary retinitis pigmentosa
Results
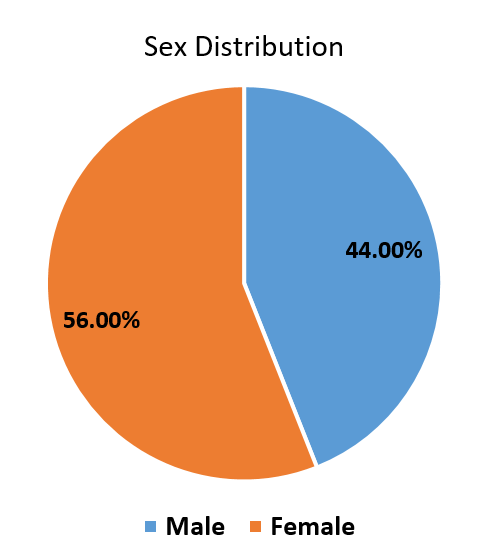
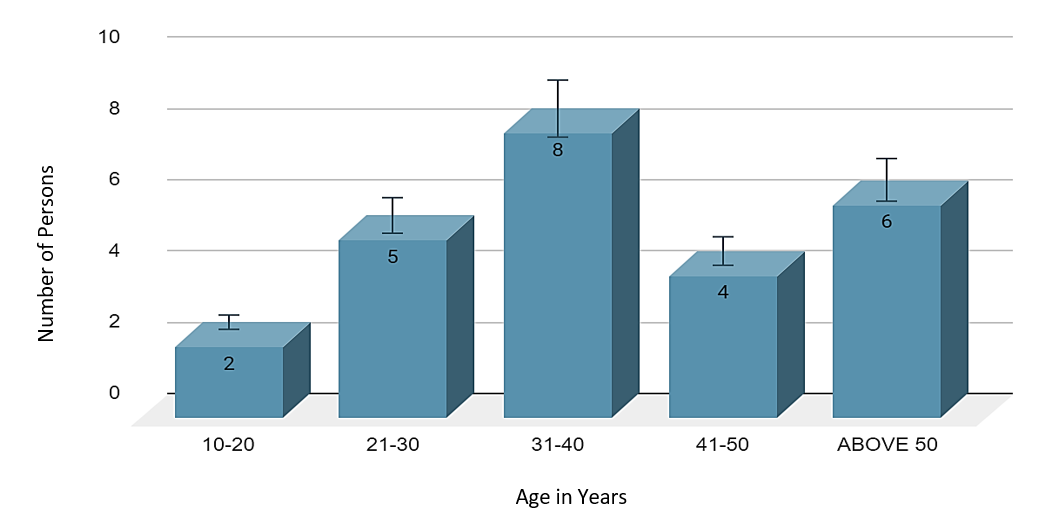
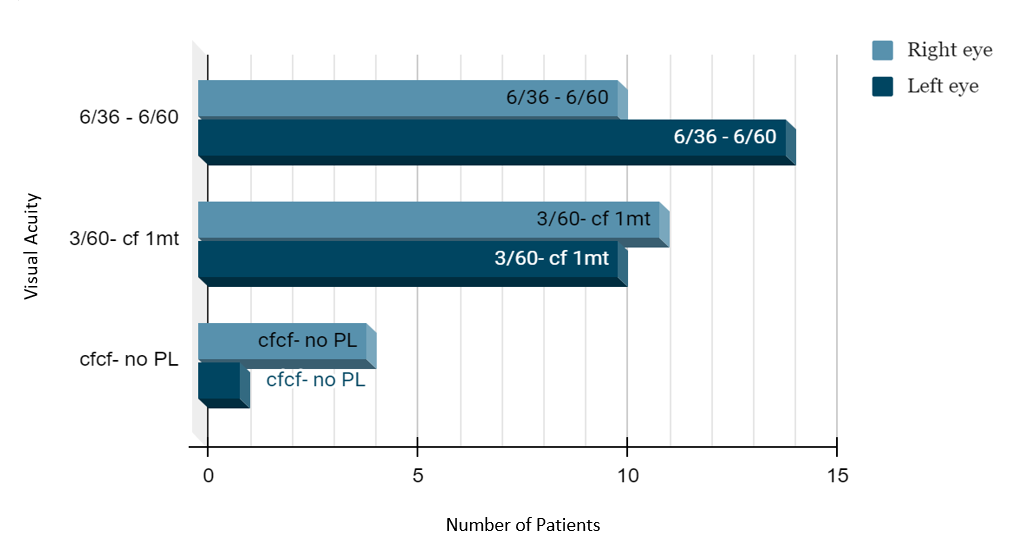
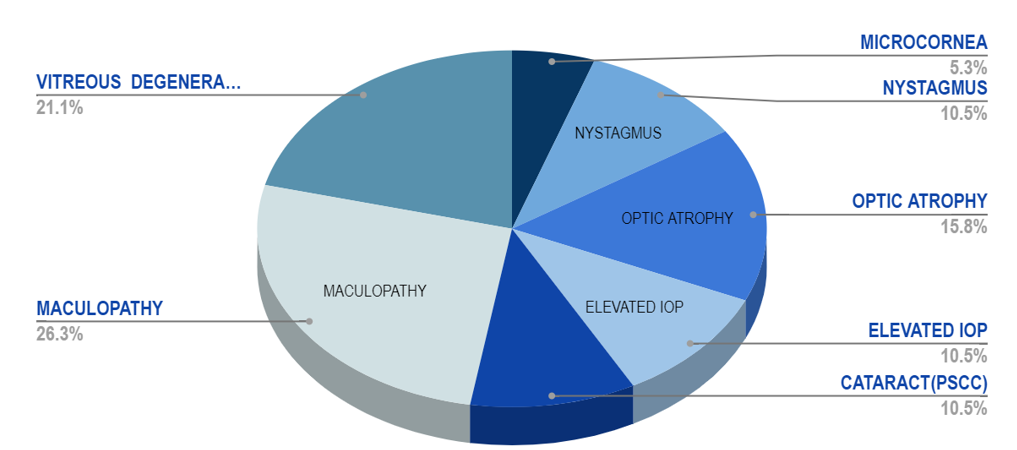
A total of 50 eyes in 25 patients were investigated, ([Figure 2]) which showed 56% females and 44% males,([Figure 1]) 40% had a history of consanguinous marriage, positive family history of RP was noted in 20%. Night blindness (58.2%) and blurred vision (27.1%) were the main visual complaints. The BCVA showed that 03 patients had complete loss of vision, and 22 had low vision, ([Figure 3]) among ocular findings most common ([Figure 5]) was maculopathy (26.3%) followed by vitreous degenerations other findings like nystagmus, microcornea, optic atrophy and posterior subcapsular cataract (most common anterior segment finding) were also present.([Figure 4])
In our study 10 patients were prescribed low vision aids which included hand held magnifiers and stand – mounted magnifiers.
Syndromic retinitis pigmentosa
In our study, Bardet biedl syndrome was found in 4 patients. The 5 cardinal features are pigmentary retinal degenerations, polydactyly, congenital obesity, mental retardation, hypogonadism.([Figure 7]) Incomplete bardet biedl syndrome is a rule rather than exception in this syndrome. Prosperi et al, estimated from previous reports that 45% of cases are incomplete which correlated with our study results. Mutations in BBS1 to BBS18 gene accounts for 70 to 80%.[3]


In our study 40% patients had history of consangious marriage, 20% patients had positive family history, for these patients pedigree chart was made and it was found that autosomal dominant trait was the most common mode of inheritance, and genetic councelling was given to these patients.
Discussion
In our study prelevance of maculopathy 26% is the most common ocular association with RP, this is in accordance with the other studies like Natarajan et al.[4] According to Campo et al. visual field loss with strong bilateral symmetry starts with solitary scotomas in the mid-peripheral zones and progresses to partial or complete ring scotomas.[5] ([Figure 8]) According to Auffarth et al., Heckenlively, and Pruett, around 45% of RP patients experience posterior subcapsular cataract, in our study 10% had PSCC.[6], [7], [8]
Cataract surgery in RP patients may result in advancement of outer layer macular retinal atrophy, macular edema, phototoxicity, posterior capsular opacification, and anterior capsular phimosis. Visual acuity does increase after cataract surgery, and there is a higher risk of posterior capsular opacification than in normal eyes, but the frequency of macular edema is reduced. Low-vision devices can be very useful to RP patients. Dark-adaptation difficulties can be overcome by using a simple penlight for searching in dark cabinets or finding a keyhole at night, for reading and writing near visual aids such as lighted magnifiers and closed-circuit televisions are useful.[9]
A major double-masked, randomized experiment found that there was no change in the visual field or visual acuity in patients who took long-term daily oral vitamin A (15,000 IU/day) compared to controls. A diet rich in omega-3 fatty acids, including docosahexaenoic acid, can further decrease disease progression. Researchers determined that the combination of vitamin A and this diet could provide about 20 years of functional eyesight for those who begin the program in their thirties. This is still a topic of debate.[10]
Retinal implants are being developed and implanted at various locations across the world. There are two types of implants: epiretinal and subretinal. The main premise is to implant microchip electrodes on or beneath the retina. They are activated by light and transform it into electrical signals. These electric impulses then stimulate biological visual signals in the remaining functional retinal cells, which are relayed to the brain via the optic nerve.[11]
Gene therapy involves replacing faulty genes with functioning ones. The most promising strategy would include employing vectors such as recombinant adeno-associated virus to transfer new genes to the retina, but there are worries about the possibility of problems when a virus is injected into the eye, as well as the safety of vectors.[12]
From our study we see most of the patients had history of consanguious marriage and positive family history thus we emphasize on the importance of genetic counselling and use of low vision aids in improving the quality of life in these patients. Genetic counselling is crucial for managing RP, as it is a heritable condition. Genetic counselling is to enlighten patients about the physical, emotional, and familial implications of genetic findings related to RP.[13] Genetic counselling needs to be tailored to the needs and profile of the patient. It involves informing patients of the hereditary nature of their disease, the prognosis and management and the risk of the disease expressing itself in other family members.[14] The diagnostic rates of genetic testing have improved due to the advent of Next Generation Sequencing (NGS) testing techniques, which have led to more personalized counselling and more accurate estimates of recurrence risks.[15] Genetic counselling includes psychological guidance for patients considering presymptomatic testing, as well as assessing the societal implications. Patients with RP may face challenges in making educated decisions about their education, career, and lifestyle. If there is a higher risk of having afflicted offspring, preconception and pre-implantation counselling should be considered.[16]
Conclusion
Mostly up to 25% of patients with RP become legally blind in both eyes, in our study 03 patients had 100% visual impairment with a visual acuity of less than 3/60. Half or even more of patients have visual acuity of 6/12 or better in at least one eye. In our study 10 patients were prescribed low vision aids which included hand held magnifiers and stand – mounted magnifiers. Thus from our study we bring to light the common ocular and syndromic associations of retinitis pigmentosa to provide awareness about the disease and genetic counselling and its benefits to the patient and their families and the use of low vision aids to improving the quality of life. There is no known prescription practice cure for the disease at present, we can help in rehablitation of such patients and prevent the disease by proper genetic counselling.
Source of Funding
None.
Conflict of Interest
None.
References
- F Testa, S Rossi, R Colucci, B Gallo, V DiIorio, MD Corte. Macular abnormalities in Italian patients with retinitis pigmentosa. Br J Ophthalmol 2014. [Google Scholar]
- OH Onakpoya, CO Adeoti, TS Oluleye, I A Ajayi, T Majengbasan, OK Olorundare. Clinical presentation and visual status of retinitis pigmentosa patients: a multicenter study in southwestern Nigeria. Clin Ophthalmol 2016. [Google Scholar]
- L Tan, Y Long, Z Li, X Ying, Jiayun Ren, C Sun. Ocular abnormalities in a large patient cohort with retinitis pigmentosa in Western China. BMC Ophthalmol 2021. [Google Scholar]
- S Natarajan. Retinitis pigmentosa: A brief overview. Indian J Ophthalmol 2011. [Google Scholar]
- RV Campo, TM Aaberg. Ocular and Systemic Manifestations of the Bardet-Biedl Syndrome. Am J Ophthalmol 1982. [Google Scholar]
- GU Auffarth, U Faller, MR Tetz, H Krastel, HE Völcker. Development of a standardized evaluation system for cataracta complicata in retinitis pigmentosa. Ophthalmologe 1997. [Google Scholar]
- J Heckenlively. The frequency of posterior subcapsular cataract in the hereditary retinal degenerations. Am J Ophthalmol 1982. [Google Scholar]
- RC Pruett. Retinitis pigmentosa: clinical observations and correlations. Trans Am Ophthalmol Soc 1983. [Google Scholar]
- C Hamel. Retinitis pigmentosa. Orphanet J Rare Dis 2006. [Google Scholar]
- XTA Nguyen, L Moekotte, AS Plomp, AA Bergen, MMv Genderen, CJF Boon. Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies. Int J Mol Sci 2023. [Google Scholar]
- N Cross, CV Steen, Y Zegaoui, A Satherley, L Angelillo. Retinitis Pigmentosa: Burden of Disease and Current Unmet Needs. Clin Ophthalmol 2022. [Google Scholar]
- C Méjécase, S Malka, Z Guan, A Slater, G Arno, M Moosajee. Practical guide to genetic screening for inherited eye diseases. Ther Adv Ophthalmol 2020. [Google Scholar]
- R Resta, BB Biesecker, RL Bennett, S Blum, SE Hahn, MN Strecker. A New Definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force Report. J. Genet. Couns 2006. [Google Scholar]
- K Shintani, DL Shechtman, AS Gurwood. Review and update: Current treatment trends for patients with retinitis pigmentosa. Optometry 2009. [Google Scholar]
- S Strait, R Loman, L Erickson, M Debenedictis. Inherited retinal degeneration current genetics practices - a needs assessment. Ophthalmic Genet 2020. [Google Scholar]
- XT Nguyen, L Moekotte, AS Plomp, AA Bergen, MMV Genderen, CJF Boon. Retinitis Pigmentosa: Current Clinical Management and Emerging Therapies. Int J Mol Sci 2023. [Google Scholar]